Dementia diagnosis: Case presentations and neuropathology
Issue: BCMJ,
vol. 47 , No. 9 , November 2005 ,
Pages 480-486 Clinical Articles
There have been significant advances in our understanding of the pathogenesis of dementia in the past two decades. We now realize that not all dementia is due to Alzheimer disease. Several new clinicopathological entities have been described and there is growing recognition that many cases of dementia have more than one underlying pathology. The specific underlying pathology or pathologies may influence the clinical manifestations, course of disease, genetic risk to family members, and responses to therapy. Longitudinal clinical evaluations of patients followed by accurate pathological diagnosis at autopsy are allowing us to better understand the syndromes and develop optimal management strategies. Three cases of patients seen at the Clinic for Alzheimer Disease and Related Disorders at UBC Hospital illustrate this.
While Alzheimer disease is a common cause of dementia, more than one underlying pathology is often involved when an individual experiences the loss of cognitive function.
Mr G. was a 74-year-old retired car salesman who was first seen at the clinic in May 1998. At that time he complained of declining memory for the past 3 years, said he had lost interest in playing golf, seemed to have lost his sense of direction, and was less energetic in activities. He was sleeping more, with prolonged daytime naps.
His medical history included a triple-vessel coronary artery bypass graft in 1989. In 1997 he was diagnosed with sinus bradycardia and hypertension. His medications included ASA (325 mg daily) and acebutolol (50 mg twice a day). He was a former smoker and had previously experienced one episode of vertigo, nausea, and vomiting with no specific diagnosis. He had a depressed mood at times. Physical examination was unremarkable and laboratory values were normal.
In May 1996 he had a CT head scan that showed normal ventricular size, normal sulci, and a tortuous basilar artery. In June 1998 a repeat CT head scan showed mild cerebral atrophy with normal ventricles but widened frontal cortical sulci and sylvian fissures, and bilateral calcification of the globus pallidus.
Prior to the assessment he had been prescribed donepezil for 5 weeks, during which time he was more sleepy and depressed. Both he and his family were convinced that the side effects from the medication were more prominent than the perceived benefit and so it was discontinued.
Six months later Mr G. became agitated and was prescribed risperidone (0.5 mg). In May 1999 he had auditory hallucinations, delusions (he couldn’t take a shower because someone was in the shower stall), utilization behavior (he took shoes out of the closet and spread them around the bedroom), and effort angina. His medications now included acebutolol, paroxetine, and nitroglycerin.
Serial mental status evaluations (Table 1) were conducted using the Mini-Mental State Examination (MMSE)[1] and the Clock Drawing Test (CDT).[2] There was a progressive decline in Mr G.’s test scores, with relative preservation of recall and the ability to spell “world” backwards. His clock-drawing ability showed significant worsening, consistent with his decline in visual-spatial problem solving—an atypical feature for AD (see educational point 2).
In August 1999 he was hospitalized with congestive heart failure and atrial fibrillation. That November he was admitted to an intermediate care facility with diagnoses of “Alzheimer disease, multiple falls, history TIAs, pneumonia, aggressive behavior.” He died in October 2001.
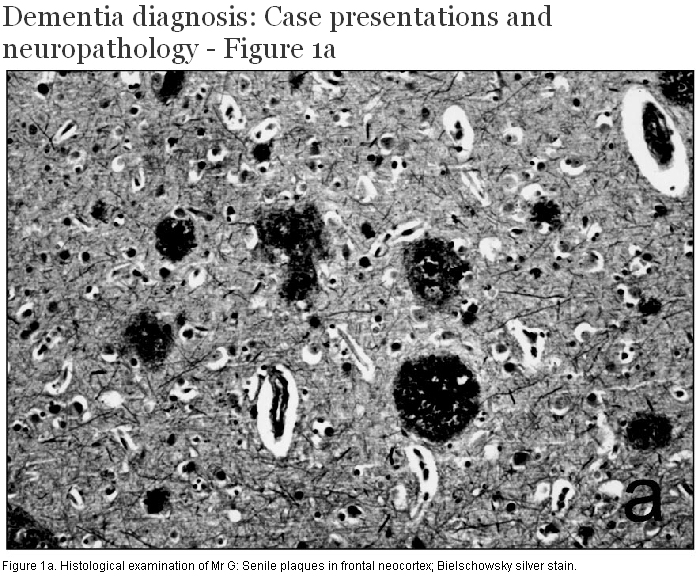
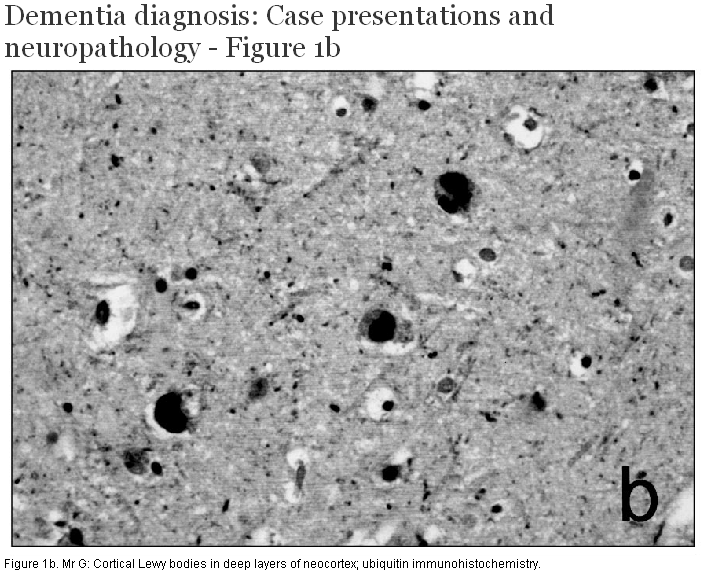
At post mortem, brain weight was 1320 g and external examination demonstrated mild atrophy of the anterior portion of the cerebral hemispheres. There was loss of pigmentation of the substantia nigra and locus ceruleus. No other focal gross lesions were identified. Histological examination showed numerous senile neuritic plaques (Figure 1a, 1b, 1c) and neurofibrillary tangles throughout the limbic system and neocortex, sufficient to fulfill current diagnostic criteria for Alzheimer disease. There was dropout of pigmented neurons from the substantia nigra, and round eosinophilic cytoplasmic inclusions consistent with Lewy bodies were seen in many of the remaining cells. Cortical Lewy bodies were identified within small neurons of the deep layers of the limbic system and neocortex using immunohistochemistry for ubiquitin and alpha-synuclein. The Lewy body score was 10/10 according to the criteria published by McKeith and colleagues.[3] Evidence of cerebrovascular disease included the presence of cerebral amyloid angiopathy affecting both leptomeningeal and cortical vessels, arteriolosclerosis in the basal ganglia and deep hemispheric white matter, and a small number of chronic microinfarcts in the neocortex and basal ganglia.
Neuropathological diagnosis: Caused by dementia (1) Alzheimer disease, (2) diffuse Lewy body disease, and (3) cerebrovascular disease.
1. It is not uncommon to have more than one pathology contributing to the dementia syndrome. The pathological substrate for this patient’s dementia was likely a combination of AD and diffuse Lewy body disease, with vascular lesions probably making a more minor contribution.
2. Even when there is a history of vascular disease (significant cardiac disease, hypertension), one cannot assume that it is the sole underlying cause of dementia.
3. Many cases that turn out to have diffuse Lewy body disease pathology do not satisfy current clinical diagnostic criteria. Lewy body dementia could have been suspected in this patient because of his difficulty with visual-spatial problem solving, agitation, psychotic features, and sensitivity to psychotropic medication.
4. Each of the underlying pathologies has potential therapeutic implications. In light of the clinical diagnosis of AD, it was appropriate to try cholinesterase inhibitor (ChEI) therapy. At the time, donepezil was the only option and unfortunately it was poorly tolerated. Nonetheless, another ChEI should have been tried since individuals with Lewy body dementia are likely to respond well to this form of treatment.[4-6] The attempt made to optimize management of vascular risk factors was suitable.
Mrs S. was an 86-year-old widowed former school teacher who was first seen at the clinic in 1998. Her daughters noted that she had decreased short-term memory, was less fastidious with her personal hygiene, was quieter, and had increased difficulty making decisions. She complained of being more tired and “lazy” but continued to do crossword puzzles.
Her medications included donepezil (5 mg daily), ASA (81 mg), and vitamin E (400 IU). On enquiry, she had a mildly unstable gait, mild back pain, and a history of periodic postural dizziness. General physical and neurological examination revealed nothing unusual and laboratory study results were normal. A CT head scan showed mild ventricular dilatation, sulcal widening, and possible small infarcts in the left caudate and putamen.
The clinical diagnosis was AD and her donepezil was increased to 10 mg daily, while her other medications were left unchanged. Review of her living situation was undertaken and suggestions were discussed with her daughters in order to streamline day-to-day supervision of medications, grooming, and nutrition.
By 2000 she had given up her apartment and was living with her daughters on rotation. In addition, she was attending an adult day centre. She was admitted to a care facility and subsequently developed atrial fibrillation and congestive heart failure. She collapsed and died 5 months after admission at age 90. Her cognition scores from tests during the last 3 years of her life (Table 2), showed relatively little change.
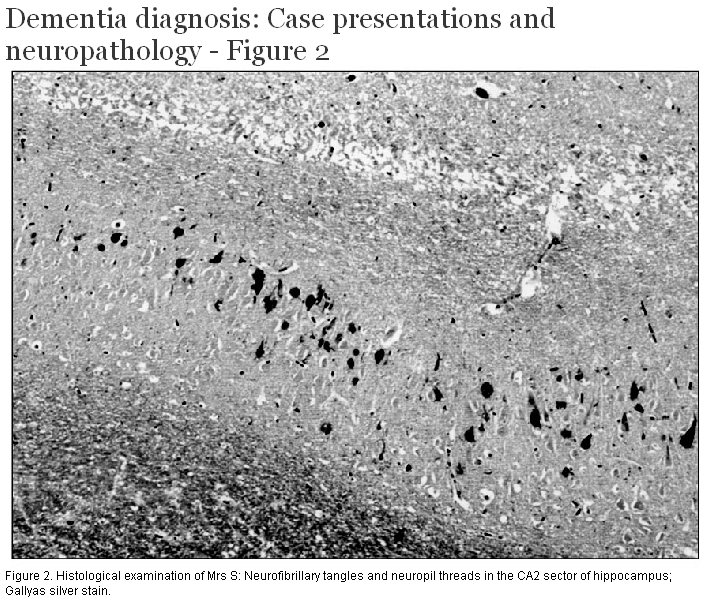
External examination of the brain demonstrated mild atrophy of the anterior portion of the cerebral hemispheres. The major histological finding was the presence of numerous silver-positive, tau-immunoreactive neurofibrillary tangles (Figure 2) and neuropil threads within the hippocampus and subiculum, and the entorhinal and transentorhinal cortex. Rare neurofibrillary tangles were present in the temporal neocortex but there were no senile neuritic plaques. The only other significant finding was the presence of numerous small round or coma-shaped argyrophilic structures in the hippocampus and subiculum, consistent in appearance with argyrophilic grains.
Neuropathological diagnosis: Neurofibrillary tangle predominant dementia.
1. Tangle-predominant dementia is a recognizable clinicopathological entity with uncertain relationship to AD. “Senile dementia with tangles” was first described by Ulrich[7] and later referred to as a “neurofibrillary tangle predominant form of senile dementia” by Brancher and Jellinger.[8] It is a sporadic disease that most often affects very old female subjects, causing mild to moderate, slowly progressive dementia. It is characterized pathologically by numerous neurofibrillary tangles that are largely restricted to the allocortex of the inferomedial temporal lobe. There is little pathology in the neocortex and senile plaques are notably absent in most cases. The incidence of this subtype of dementia ranges from 0.7% to 7.7% in several autopsy series and reached 8.3% and 10.2% in two series of centenarians. Progression of the dementia is slow and usually meets criteria for probable or possible AD as defined by the National Institute of Neurological and Communication Disorders and Stroke and Alzheimer Disease and Related Disorders Association.[9] Death usually occurs after age 85. A few patients have extrapyramidal signs, including rigidity and gait disorder.
2. It was only through neuropathological evaluation that the true diagnosis was revealed. Although the clinical course of this patient was compatible with tangle-predominant dementia, this uncommon condition was not considered.
3. Donepezil therapy in many cases of AD appears to keep people stable over time.[10] The effect of this medication in tangle-predominant dementia is unknown. In this case, the course of the disease was slow, but the patient had more functional decline than her cognitive scores would indicate, underlining the complexity of evaluating the dementia syndrome.
Mr N. was a 71-year-old carpenter who had a rather abrupt change in his personality when his brother died in March 1999. He began to cry easily and frequently. At times he stared into space, was repetitive in conversation, and was paranoid about break-ins to the house and car. He was short-tempered and had difficulty with words (e.g., calling the supermarket “the thing”). He continued to downhill ski without difficulty. Family history was unremarkable.
Mr N. was first seen in the clinic in 2000. In addition to describing the symptoms listed above, his wife indicated he had become sexually disinhibited. She also indicated he never stopped being active and described him as “go, go, go.” His family physician prescribed venlafaxine and donepezil.
General physical and neurological examination results were normal. Laboratory study results were also normal. A CT head scan showed mild frontotemporal atrophy and a SPECT scan showed decreased frontal perfusion.
In 2001 he became more disinhibited. There was significant decrease in communication with occasional single words in his first language, frequent smiling, and occasional inappropriate laughing. By the late fall, his wife reported that he was developing significant difficulty with swallowing and that he was coughing frequently. She altered his diet to thickened fluids. He remained active, continuing to walk every day until his final hospital admission for aspiration pneumonia. He died shortly after admission in April 2002, approximately 3 years after onset of symptoms.
When Mr N. was last seen at the clinic in September 2001, he was unable to do the MMSE because of his language difficulties and the Severe Impairment Battery (SIB)[11] was administered instead. Even on this test, which tends to be used when the MMSE score is 11 or less, he was very impaired (Table 3).
At post mortem, brain weight was 1396 g and there was moderate cerebral atrophy, which predominantly affected the frontal and anterior temporal lobes. Histological sections showed a linear band of spongiotic change affecting the superficial neocortex. No senile neuritic plaques or neurofibrillary tangles were identified. Ubiquitin imunohistochemistry demonstrated neuropil threads and small, round cytoplasmic neuronal inclusions in the superficial layers of the neocortex and cytoplasmic inclusions in the dentate granule cells of the hippocampus (Figure 3a, 3b). Although the number of lower motor neurons in the brainstem appeared to be normal, some contained cytoplasmic inclusions with the morphology of filamentous skeins or dense round bodies immunoreactive for ubiquitin. The spinal cord was not available for examination.
Neuropathological diagnosis: Frontotemporal dementia (FTD) with motor neuron disease (MND) type inclusions.
1. Frontotemporal dementia is a recognizable clinical syndrome characterized by changes in personality, behavior, and speech disturbance.[12] FTD may be accompanied by pyramidal or extrapyramidal features. This patient developed features that may have represented early pyramidal system dysfunction.
2. Several different patterns of pathology may underlie FTD.[13] In almost half of patients, the pathology is characterized by the abnormal accumulation of tau protein in neurons and glia. These so-called tauopathies include classical Pick disease, corticobasal degeneration, progressive supranuclear palsy, and FTD with parkinsonism linked to chromosome 17 (FTDP-17). More commonly, cases of FTD are found to have no tau pathology, but instead have neuronal cytoplasmic inclusions that are ubiquitin-immunoreactive (FTD-U) (Figure 3a, 3b). This FTD-U pattern of pathology is a consistent finding in patients with motor neuron disease and dementia, and is also found in some patients with FTD in the absence of motor symptoms (referred to as FTD-MND-type or MND inclusion dementia). The finding of inclusions in motor neurons in some patients with FTD-MND-type (such as ours) suggests that MND, MND with dementia, and FTD-MND-type dementia represent a clinicopathological spectrum of disease.
3. The specific pathology underlying FTD can only be determined through neuropathological examination.
4. At present there are no specific treatment strategies for FTD and management is directed at prevention of aspiration and symptomatic care. There is no evidence that ChEIs are helpful. In some cases of FTD there is a strong genetic influence and genetic counseling may be helpful to surviving family members.
The three cases presented here show that a large number of conditions may cause dementia. Although Alzheimer disease is the likely cause in the majority of cases, less common conditions such as tangle-predominant dementia and more recently recognized clinicopathological entities such as FTD with MND-type inclusions need to be considered.
These cases also indicate that more than one type of pathology may be present in a patient with dementia. Each of the pathologies may influence the clinical features and course of disease, and may have different genetic risk factors. Moreover, each pathological process has distinct treatment implications:
• Vascular-related clinical problems (hypertension, hypercholesterolemia, and atrial fibrillation) must be recognized and modified to avoid making the dementia unnecessarily worse.
• Using psychotropic agents to treat disturbing behavior in patients with diffuse Lewy body disease may produce agitation, psychotic features, and parkinsonism, so these agents must be prescribed with caution.
• Although there are symptomatic treatments for dementia, they don’t work in all patients to the same degree. Those patients with AD may have mild improvement or appear to stay the same longer. Those with diffuse Lewy body disease may have better responses to cholinesterase inhibitors. And those with FTD do not appear to respond to this form of treatment.
• There is no evidence that ChEIs are effective for tangle-only dementia, which in many cases may be a very slowly progressive disorder. The disorder is not found frequently and is difficult to distinguish clinically from AD.
• Because dementia is a syndrome with cognitive, functional, and behavioral aspects, each component requires attention with a combination of nonpharmacological and pharmacological interventions and close follow-up.
Finally, these cases show that the specific type of pathology in a patient with dementia can only be determined by neuropathological examination. Postmortem evaluation of patients with neurodegenerative diseases not only confirms diagnosis but provides valuable insight into the effectiveness of various treatment strategies and allows for appropriate genetic counseling of family members.
Competing interests
Dr Beattie has been a co-investigator for pharmaceutical companies and participated in CME programs, but is not a stock shareholder.
Table 1. Mental status evaluations for Mr G.
| Jan 1998 | May 1998 | Jan 1999 | May 1999 | |
| MMSE (30) | 30 | 26 | 24 | 21 |
| CDT (15) | — | 12 | 4 | 4 |
Table 2. Mental status evaluations for Mrs S.
| 1998 | 1999 | 2000 | |
| MMSE (30) | 30 | 26 | 24 |
| CDT (15) | 15 | 15 | 15 |
Table 3. Mental status evaluations for Mr N.
| Feb 2000 | Apr 2000 | Sep 2001 | |
| MMSE (30) | 24 | 25 | — |
| CDT (15) | — | 12 | 30 |
References
1. Folstein MF, Folstein SF, McHugh PR. “Mini-mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189-198. PubMed Citation Full Text
2. Shulman K, Shedletsky R, Silver I. The challenge of time: Clock drawing and cognitive function in the elderly. Int J Geriatr Psychiatry 1986;1:135-140.
3. McKeith IG, Perry RH, Fairbairn AF, et al. Operational criteria for senile dementia of Lewy Body type (SDLT). Psychol Med 1992;22:911-922. PubMed Abstract
4. Aarsland D, Mosimann UP, McKeith IG. Role of cholinesterase inhibitors in Parkinson’s disease and dementia with Lewy bodies. J Geriatr Psychiatry Neurol 2004;17:164-171. PubMed Abstract Full Text
5. Simard M, van Reekum R. The acetylcholinesterase inhibitors for treatment of cognitive and behavioral symptoms in dementia with Lewy bodies. J Neuropsychiatry Clin Neurosci 2004;16:409-425. PubMed Abstract Full Text
6. McKeith IG, Grace JB, Walker Z, et al. Rivastigmine in the treatment of dementia with Lewy bodies: Preliminary findings from an open trial. Int J Geriatr Psychiatry 2000;15:387-392. PubMed Abstract Full Text
7. Ulrich J. Histochemical representation of acetylcholinesterase in Alzheimer’s disease. Acta Histochem Suppl 1992;42:11-12. PubMed Abstract
8. Brancher C, Jellinger K. Neurofibrillary tangle predominant form of senile dementia of Alzheimer type: A rare subtype in very old subjects. Acta Neuropathol (Berl) 1994;88:565-570. PubMed Abstract
9. McKhann G, Drachman DA, Folstein M, et al. Clinical diagnosis of Alzheimer’s disease: Report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 1984;34:939-944. PubMed Abstract
10. Passmore AP, Bayer AJ, Steinhagen-Thiessen E. Cognitive, global, and functional benefits of donepezil in Alzheimer’s disease and vascular dementia: Results from large-scale clinical trials. J Neurol Sci 2005;229-230:141-146. PubMed Abstract Full Text
11. Saxton J, McGonigle-Gibson K, Swihart A, et al. Assessment of the severely impaired patient: Description and validation of a new neuropsychological test battery. Psychol Assess 1990;2:298-303.
12. Neary D, Snowden JS, Mann DM. Classification and description of frontotemporal dementias. Ann NY Acad Sci 2000;920:46-51. PubMed Abstract Full Text
13. Trojanowski JQ, Dickson D. Update on the neuropathological diagnosis of frontotemporal dementias. J Neuropathol Exp Neurol 2001;60:1123-1126. PubMed Citation Full Text
14. Okamoto K, Murakami N, Kusaka H, et al. Ubiquitin-positive intraneuronal inclusions in the extramotor cortices of presenile dementia patients with motor neuron disease. J Neurol 1992;239:426-430. PubMed Abstract
15. Jackson M, Lowe J. The new neuropathology of degenerative frontotemporal dementias. Acta Neuropathol (Berl) 1996;91:127-134. PubMed Abstract Full Text
B. Lynn Beattie, MD, FRCPC, Ian R.A. Mackenzie, MD, FRCPC
Dr Beattie is professor emerita, Geriatric Medicine at the University of British Columbia and former director of the Clinic for Alzheimer Disease and Related Disorders at UBC Hospital. Dr Mackenzie is a professor in the Department of Pathology (Neuropathology) at UBC and Vancouver General Hospital.

{kind=link}
{kind=link}
{kind=link}
{kind=link}