Validity of carotid intimal-medial thickness as a surrogate endpoint for atherosclerosis: Lessons from the ENHANCE trial
Issue: BCMJ,
vol. 51 , No. 4 , May 2009 ,
Pages 168-172 Clinical Articles
Contemporary evidence-based medicine aims to apply knowledge gleaned from the scientific method to medical practice. While clinical trials based on meaningful endpoints provide the most robust guidance, the measurement of these may be inappropriate or not technically and financially feasible in many clinical situations. As a result, clinically related, laboratory-derived surrogate endpoints have been proposed as alternatives. In the case of atherosclerosis, clinical endpoints such as stroke, myocardial infarction, and amputation are often preceded by many years of subclinical arterial wall changes and luminal narrowing. The recent ENHANCE trial used carotid intimal-medial thickness as a surrogate endpoint in its evaluation of ezetimibe, and ultimately generated more questions than answers. With numerous other trials presently underway, we are likely to obtain additional evidence about ezetimibe and its impact on atherosclerosis in future. In the meantime, it appears prudent to prescribe ezetimibe only for individuals with atherosclerosis or at sufficient risk of atherosclerosis who cannot reach the guideline-recommended level of low-density-lipoprotein cholesterol on the highest tolerated dose of statin.
While there are no robust clinical outcome data to support the use of ezetimine as a cholesterol-lowering drug, there are reasons to consider luminal narrowing as a surrogate for atherosclerosis.
In contemporary medicine, the clinician must be able to readily incorporate information from clinical trials into his or her busy clinical practice. This process is made easier when the information is derived from rigorously conducted, large-scale, randomized clinical trials with meaningful endpoints (such as trials that show a reduction in mortality from a particular intervention). However, in modern clinical trials, measuring these endpoints may be difficult or impossible. This limitation is especially true in the study of atherosclerosis.
Atherosclerosis is a dynamic process that occurs over decades. Clinical endpoints such as stroke, myocardial infarction (MI), and amputation are often preceded by many years of progressive subclinical arterial wall changes and luminal narrowing. Unfortunately, any clinical trial looking at an intervention that may alter these changes requires an extraordinarily large number of patients who need to be followed over a long period. Trials of this magnitude are not feasible technically and financially. As a result, clinically related, laboratory-derived surrogate endpoints have been proposed as alternative endpoints to be used in these situations. Intimal-medial thickness (IMT) progression has been proposed as one such surrogate endpoint in the study of atherosclerosis progression. In the recent ENHANCE trial (Effect of Combination Ezetimibe and High-Dose Simvastatin versus Simvastatin Alone on the Atherosclerotic Process in Patients with Heterozygous Familial Hypercholesterolemia),[1] IMT measurements were used to evaluate the ability of ezetimibe to halt atherosclerosis.
Clinicians can benefit from looking at the original derivation of clinical surrogate endpoints and previous research that has validated IMT as a surrogate endpoint. They can also benefit from considering how the findings about ezetimibe from the ENHANCE trial might be applied to clinical practice.
Surrogate endpoints
A surrogate endpoint is an intermediate variable that is statistically related to the clinical endpoint of interest.[2] Surrogate endpoints have been used in numerous recent clinical trials to save time and money, and to make studies with interventions on longitudinal disease processes feasible. The stronger the relationship between a surrogate endpoint and the clinical endpoint of interest, the better a clinician is able to interpret changes in the surrogate endpoint as clinically meaningful. Common examples of surrogate endpoints include the measurement of glycosylated hemoglobin (HbA1c) as a surrogate for long-term diabetes control and cholesterol or blood pressure reduction as a surrogate endpoint for reductions in cardiovascular (CV) events or mortality.
In order to assess the clinical validity of a surrogate endpoint, Boissel and colleagues[2] described three requirements that must be met. The first requirement is that the surrogate endpoint should occur more frequently than the clinical endpoint and therefore be much easier to assess. The second requirement is that the surrogate endpoint should have a relationship to the clinical endpoint, and this relationship needs arise from epidemiological data. The third requirement is that the surrogate endpoint must accurately provide an “estimate of clinical benefit.” Thus, by modifying the surrogate endpoint, one should be able to estimate a similar change in the clinical endpoint of interest. This is the most important criterion, as it allows us to deduce changes in the clinical endpoint through changes in the surrogate endpoint. This estimation is usually obtained through clinical trial data.
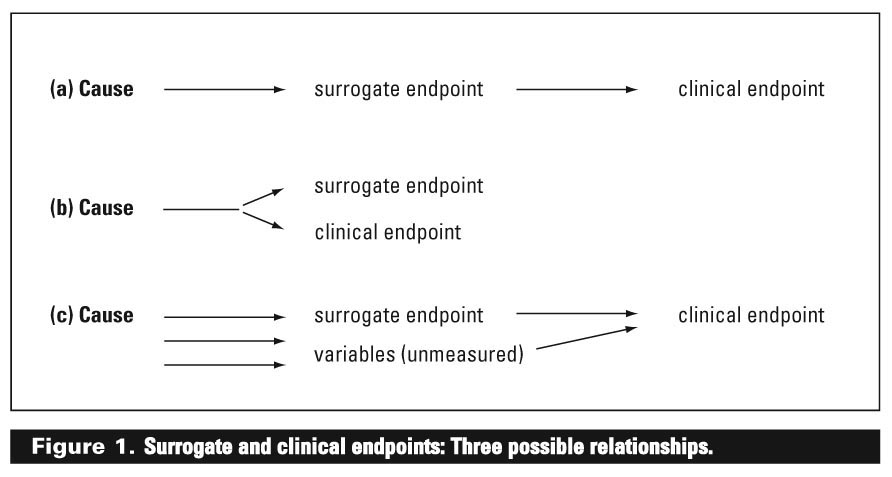
It is important to keep in mind that the relationship between the surrogate endpoint and the clinical endpoint may not be direct. There are numerous relationships that may exist. Three possible relationships are shown in Figure 1.
{kind=link}
In the first (a), the relationship is direct, as the surrogate endpoint is an intermediate event or condition on the pathway leading to the clinical endpoint of interest. One such example would be measuring beta-hydroxybutyric acid as a surrogate for ketone production in patients with suspected diabetic ketoacidosis. In this example, the surrogate endpoint would be an ideal one to measure because measuring the surrogate endpoint would provide direct information about the clinical endpoint of interest.
In the second relationship (b), the surrogate endpoint and the clinical endpoint share a remote cause. Although the clinical endpoint of interest cannot occur without the surrogate endpoint occurring, the surrogate endpoint does not cause the clinical endpoint. An example of this would be measurement of HbA1c as a surrogate endpoint of cellular glycemic control. In this case, both share a common cause (persistent hyperglycemia), but HbA1c does not cause cellular glycemic control. In this situation, the surrogate endpoint would also be an ideal one to measure because the clinical endpoint cannot occur without the surrogate endpoint occurring.
In the third relationship (c), the clinical endpoint of interest may have multiple causes, only one of which causes the surrogate endpoint. In this situation, the surrogate endpoint has a complex relationship with the clinical endpoint of interest. It may, therefore, not be an ideal surrogate endpoint, and changes in the surrogate endpoint may not adequately reflect changes in the clinical endpoint of interest. An example of this would be using homocysteine levels as a surrogate endpoint for cardiovascular events.
Intimal-medial thickness as a surrogate endpoint
Atherosclerosis is a complex process leading to the progressive narrowing of arterial lumina. A preliminary common pathway of this process appears to be thickening of the arterial walls. Using B-mode ultrasonography (see Figure 2), the combined thickness of the intima and media can be assessed (as measured from the inner intimal layer to the outer medial layer of the wall). It is deduced that changes in IMT reflect atherosclerotic changes. Unfortunately, other disease process can also cause changes in medial thickness (such as infiltration or infection of medial layers). IMT measurements cannot distinguish between these changes and atherosclerotic thickening, and consequently may lead to overestimation of atherosclerotic changes. For this reason, multiple samples of different segments are taken since the global change in IMT is more likely to reflect atherosclerotic changes than IMT changes in specific segments.
Pignoli and colleagues were the first to introduce IMT as a way to measure wall atherosclerosis.[3]In their study, the authors correlated IMT ultrasound measurements to gross pathological specimens from normal and atherosclerotic arterial samples of 10 young male patients. They concluded that IMT measurements did not differ significantly between modalities; however, there was a greater degree of error in samples with established atherosclerotic plaques (approximately 10% to 20%). They concluded that IMT measurements using ultrasound are best undertaken in the early stages of atherosclerotic disease.
Following the introduction of ultrasonography for measuring IMT, there have been a number of studies that have attempted to validate IMT progression as a surrogate endpoint for atherosclerosis progression.[4] The first epidemiological data came from two large cohort studies: the Rotterdam Study and the Atherosclerosis Risk in Communities (ARIC) Study.[5,6] In the Rotterdam Study,[5] the risk of MI increased by approximately 40% per standard deviation increase in IMT measurements, with the highest risk being seen in patients with an IMT measurement of greater than 0.908 mm. In the ARIC study,[6,7] every 0.2 mm progression in IMT measurements was associated with a 28% relative increase in the risk of stroke and a 33% relative increase in the risk of MI. This risk was highest in patients with established CV risk factors, such as hypertension and smoking. These studies provided the first clinical data suggesting that increases in IMT measurements were associated with an increase in CV events, thereby making IMT progression a possible surrogate for atherosclerosis progression.
Since these two large epidemiological studies, there have been several prospective trials that have examined the effect of various lipid-lowering strategies on IMT progression/regression and CV endpoints. The first such trial was the Cholesterol Lowering Atherosclerosis Study (CLAS),[8] a randomized, placebo-controlled angiographic study that examined the effect of colestipol-niacin therapy in nonsmoking men with progressive atherosclerosis and previous coronary artery bypass surgery. Carotid IMT ultrasound measurements were taken in a subset of 78 individuals and repeated at 2 and 4 years. The authors demonstrated a progressive reduction in carotid IMT in patients on colestipol-niacin, with a corresponding improvement in lipid levels. Though the study was not intended to look for CV endpoints, it was the first interventional trial to use IMT progression as a surrogate for atherosclerosis progression.
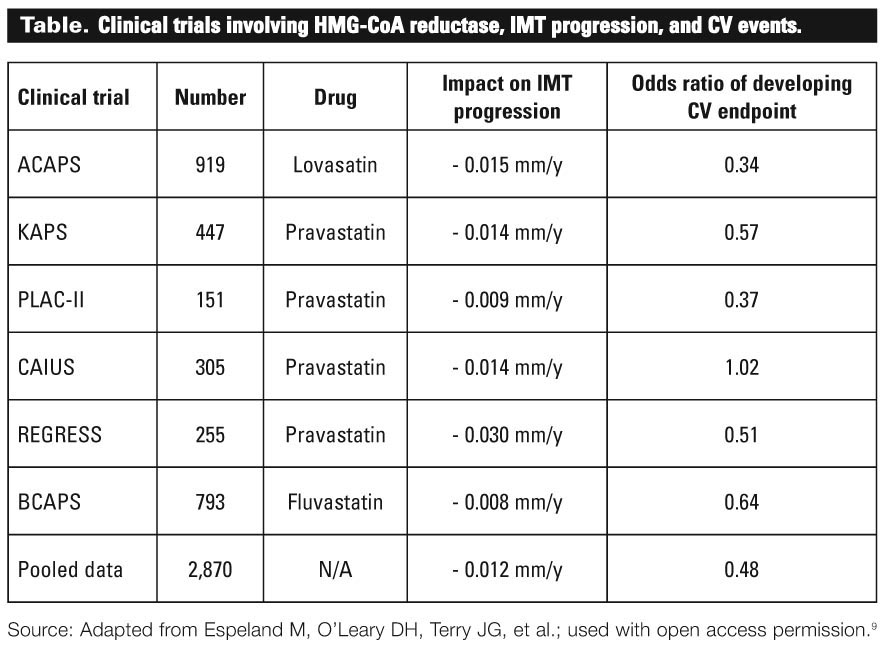
Following CLAS were a number of studies looking at the role of HMG-CoA reductase (statin) therapy with respect to IMT progression. These trials were added to a recent meta-analysis by Espeland and colleagues[9] that evaluated the rates of IMT progression and corresponding rates of CV endpoints from six large clinical trials involving nearly 3000 patients on various statin treatments (see the Table). The pooled results demonstrated an overall decrease in mean IMT progression of 0.012 mm/y in patients on the treatment group and a corresponding odds ratio of developing CV endpoint (such as CV death, MI, and stroke) of 0.48. Overall, the results of these trials suggest that halting IMT progression can significantly reduce CV endpoints. Furthermore, these studies suggest that reductions in the rates of atherosclerosis progression can be deduced through reduction in rates of IMT progression.
{kind=link}
Ezetimibe and the ENHANCE story
In vivo research first suggested that Neimann-Pick C1 Like 1 (NPC1L1) protein located on the intestinal brush border was essential for uptake of cholesterol and non-cholesterol sterols.[10] This led to the development of ezetimibe, a selective inhibitor of the NPC1L1 protein. In a 12-week pilot study, Ballantyne and colleagues[11] examined the effect of the addition of ezetimibe versus placebo to atorvastatin in patients with established dyslipidemia. With the addition of ezetimibe, the authors were able to show an additional 12% to 15% reduction in low-density-lipoprotein cholesterol (LDL-C) levels.
These initial promising results led to the ENHANCE study,1 which was intended to look at IMT progression on a select high-risk group of 720 patients with familial hypercholesterolemia treated with simvastatin plus ezetimibe versus placebo. Over the course of the 24-week study, there was a 20% further reduction in LDL-C and total cholesterol in the combination therapy group compared with the simvastatin monotherapy group. However, there was no appreciable difference in IMT progression, though both groups had essentially halted IMT progression. The study also did not show significant differences in adverse events between the groups, and was not powered to look at differences in CV endpoints. Subsequently, the trial was considered a failure. Critics questioned the methodology of the study and challenged the role of IMT progression as an adequate surrogate of atherosclerosis progression. Was the previous data linking IMT progression to atherosclerosis progression wrong? And what was to be done about ezetimibe? Should doctors continue to prescribe a drug that averaged nearly $5 billion in sales in North America in 2007 with no robust clinical outcomes?[12]
Conclusions
So how does the clinician interpret IMT studies and integrate the findings of ENHANCE into his or her practice? To interpret the IMT trials, we first have to decide whether IMT progression serves as a suitable surrogate endpoint for atherosclerosis progression. Applying the requirements proposed by Boissel and colleagues,[2] IMT measurement is convenient (requirement 1) and clearly linked to atherosclerosis progression from epidemiological data (requirement 2). From clinical studies, there is considerable evidence to support that halting IMT progression has a favorable impact on cardiovascular endpoints (requirement 3). The initial work also reminds us that IMT measurements were most accurate in study subjects with early atherosclerotic changes, and significant errors began to occur when the measurements were applied to specimens with established atherosclerotic plaques.
If we examine the results of the ENHANCE trial in this light, some unique criticisms become clear. First, the population used in the ENHANCE study had familial hypercholesterolemia—a genetic disorder associated with advanced atherosclerosis and premature cardiovascular disease. Most of these patients had previously received cholesterol-lowering drugs. Given the advanced atherosclerotic burden, it is quite possible that IMT measurements were not the ideal modality to use when monitoring atherosclerosis progression in this group. Also, the trial used only a 6-week washout period, in which patients were taken off their previous lipid-lowering drugs.[13] This was possibly insufficient time and could have confounded the results. Next, the combination group derived only an additional 20% reduction in LDL-C and total cholesterol compared with the simvastatin monotherapy group. Based on previous research we know that there is little observed benefit in CV endpoints until there is at least a 30% reduction in LDL-C levels.[14] Perhaps the degree of LDL-C reduction is the key factor in reductions in atherosclerosis progression. Finally, both groups demonstrated a near halting in IMT progression. As suggested earlier, perhaps this is ultimately the important variable in reducing CV endpoints.
Overall, the ENHANCE trial generated more questions than answers about ezetimibe and its clinical use in reducing atherosclerosis progression. Currently, there are no robust clinical outcome data to support its use as a cholesterol-lowering drug, and it appears prudent to use ezetimibe in the clinical situations it was first intended for—that is, in individuals with or at sufficient risk for atherosclerosis who cannot reach the guideline-recommended level of LDL-C on the highest tolerated dose of statin. There are, however, numerous trials presently underway looking at the effect of ezetimibe on objective clinical outcomes. We are likely to learn much more about this drug and its potential impact on atherosclerosis in the future.
When interpreting results derived from clinical trials using surrogate endpoints, it is helpful to ask questions based on the requirements outlined by Boissel and colleagues: 1. Does the surrogate endpoint occur more frequently than the clinical endpoint? 2. Does epidemiological data indicate that there is a relationship between the surrogate and clinical endpoints? 3. Does the surrogate endpoint provide an estimate of clinical benefit? It is also helpful to maintain a healthy sense of skepticism for interventions that do not have robust clinical outcome data supporting their use: our health system and the patients we care for deserve as much.
Competing interests
None declared.
References
1. Kastelein J, Akdim F, Stroes ES, et al. Simvastatin with or without ezetimibe in familal hypercholesterolemia. N Engl J Med 2008;358:1431-1443. PubMed Abstract Full Text
2. Boissel JP, Collet JP, Moleur P, et al. Surrogate endpoints: A basis for a rational approach. Eur J Clin Pharmacol 1992;43:235-244. PubMed Abstract
3. Pignoli P, Tremoli E, Poli A, et al. Intimal medial thickness of arterial wall: A direct measurement with ultrasound imaging. Circulation 1986;74:1399-1406. PubMed Abstract Full Text
4. de Groot E, Hovingh GK, Wiegman A, et al. Measurement of arterial wall thickness as a surrogate marker for atherosclerosis. Circulation 2004;109(23 suppl 1): III33-III38. PubMed Abstract Full Text
5. Bots M, Hoes AW, Koudstoal PJ, et al. Common carotid intima-media thickness and risk of stroke and myocardial infarction: The Rotterdam Study. Circulation 1997;96:1432-1461. PubMed Abstract Full Text
6. Heiss G Sharrett AR, Barnes R, et al. Carotid atherosclerosis measured by B-mode ultrasound in populations: Associations with cardiovascular risk factors in the ARIC Study. Am J Epidemiol 1991;143:250-256. PubMed Abstract
7. ARIC Investigators. Association of coronary heart disease incidence with carotid arterial wall thickness and major risk factors: The Atherosclerosis Risk in Communities (ARIC) Study, 1987-1993. Am J Epidemiol 1997;146:483-494. PubMed Abstract Full Text
8. Blankenhorn DH, Selzer RH, Crawford DW, et al. Beneficial effects of colestipol-niacin therapy on the common carotid artery. Two- and four-year reduction of intima-media thickness measured by ultrasound. Circulation 1993;88:20-28. PubMed Abstract Full Text
9. Espeland M, O’Leary DH, Terry JG, et al. Carotid intimal-media thickness as a surrogate for cardiovascular disease events in trials of HMG-CoA reductase inhibitors. Curr Control Trials Cardiovasc Med 2005;6:3. http://www.biomedcentral.com/content/pdf/1468-6708-6-3.pdf (accessed 21 May 2009).
10. Altmann SW, Davis HR Jr, Zhu JL, et al. Niemann-PickC1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004;303:1201-1204. PubMed Abstract
11. Ballantyne CM, Houri J, Notarbartolo A, et al. Effect of ezetimibe coadministered with atorvastatin in 628 patients with primary hypercholesterolemia: A prospective, randomized, double-blind trial. Circulation 2003;107:2409-2415. PubMed Abstract Full Text
12. O’Riordan M. Questions arise about the chronology of events in the ENHANCE trial. Heartwire.org 31 January 2008. http://theheart.org/article/840879.do (accessed 6 August 2008).
13. Kastelein JJ, Sager PT, de Groot E, et al. Comparison of ezetimibe plus simvastatin versus simvastatin monotherapy on atherosclerosis progression in familial hypercholesterolemia. Design and rationale of the Ezetimibe and Simvastatin in Hypercholesterolemia Enhances Atherosclerosis Regression (ENHANCE) trial. Am Heart J 2005;149:234-239. PubMed Abstract
14. Robinson J, Smith B, Maheshwari N, et al. Pleiotropic effects of statins: Benefit beyond cholesterol reduction? A meta-regression analysis. J Am Coll Cardiol 2005;46:1855-1862. PubMed Abstract
Dr Mohamed was an adult cardiology fellow in the Division of Cardiology at the University of British Columbia. He was killed in a hit and run accident in February 2009. Dr Andrade is a cardiology fellow at UBC. Dr Ignaszewski is the head of the Division of Cardiology at St. Paul’s Hospital and medical director of the hospital’s Healthy Heart Program. He is also a clinical professor of medicine at UBC. Dr Mancini is a professor in the Department of Medicine and director of Continuing Medical Education at UBC. He is also director of the Cardiovascular Imaging Research Core Laboratory in the Division of Cardiology at Vancouver Hospital.