Soft tissue sarcomas of the extremities: How to stay out of trouble
Issue: BCMJ,
vol. 50 , No. 6 , July August 2008 ,
Pages 310-317 Clinical Articles
Although the majority of soft tissue masses are benign, it is important to consider malignancy in the differential diagnosis. Because most soft tissue sarcomas present as a painless mass, clinicians must watch for signs suggestive of malignancy, including large size, rapid growth, and location deep to the deep fascia. MRI is the standard imaging modality for investigating possibly malignant masses. Local and systemic staging are essential for prognostication and subsequent management. Following staging and a review of the imaging studies, biopsy should be performed with the assistance of a surgeon experienced in treating soft tissue sarcoma. Management must then be considered in terms of both local and systemic control. Errors in the early management of sarcoma can lead to increased rates of amputation and death, making it essential that soft tissue sarcomas be distinguished from benign soft tissue masses. Family physicians should be aware that the BC Cancer Agency soft tissue sarcoma group is willing to review cases where soft tissue sarcoma is in the differential diagnosis.
An appropriately planned and executed biopsy of a soft tissue mass can help prevent misdiagnosis, increased mortality, and lost opportunities for limb salvage surgery.
A soft tissue mass is a relatively common presenting complaint in the family physician’s office. The vast majority of these lesions are benign, but very occasionally a mass will represent a soft tissue sarcoma.
Failure to recognize a sarcoma in the primary care setting and refer a patient appropriately can result in lost opportunities for limb salvage and increased mortality for the patient.[1-4]
In 2004, 84 patients with soft tissue sarcomas were referred to the BC Cancer Agency (BCCA). Considering there are 111 family practice physicians per 100000 people in British Columbia,[5] less than 1% of family practice physicians in BC will encounter a malignant soft tissue sarcoma each year. On average, however, every family practice physician will encounter three benign soft tissue masses per year.
Improved outcomes in soft tissue sarcomas managed in a multidisciplinary environment have been well documented, and it has also been shown that patients receiving biopsies or surgical interventions prior to referral to a multidisciplinary centre have inferior outcomes.[1-4]
A multidisciplinary centre has the advantages of access to sarcoma subspecialists in the fields of surgery, radiology, pathology, radiation oncology, and medical oncology as well as experienced allied health professionals essential to patient care.
Distinguishing between the malignant lesions that require early referral and the many that do not remains a significant challenge. No specialist centre could cope with seeing every presenting lump or bump, and each centre in Canada relies on the ability of family physicians to assess these lesions appropriately.
Physicians faced with a soft tissue mass need to make safe and accurate clinical decisions about referral. They also need to understand some of the principles of management followed at the BC Cancer Agency and know about recent advances in the management of these challenging lesions in order to answer many of their patients’ questions.
Epidemiology and pathology
As soft tissue sarcomas represent only 1% of all adult malignancies, it is difficult to make general conclusions about epidemiology.[6] Soft tissue sarcomas are distributed among all age groups, and although some reports suggest an increase in incidence among men,[7] most reports suggest both genders are equally susceptible. There is no known preponderance of sarcomas related to ethnicity.
Approximately 50% of all lesions occur in the extremities, with 75% of these arising in a lower extremity;[8] however, lesions can occur in any location. The incomplete understanding of the origins of these tumors is reflected in their complex and seemingly arbitrary nomenclature.
Modern genetic techniques are unravelling these mysteries. It is a common misconception that sarcomas are named after the tissue compartment in which they arise. For example, we often see a soft tissue sarcoma in a skeletal muscle referred to as a rhabdomyosarcoma.
In fact, rhabdomyosarcomas are rare in adults and make up about one-half of all soft issue sarcomas in children. Young adults show a greater prevalence of synovial sarcomas and epithelioid sarcomas,[9] keeping in mind that synovial sarcomas have nothing to do with synovium, and indeed they are not related to joints at all.
For most subtypes of sarcoma the specific cause is unknown. However, there are some factors thought to be associated with specific sarcomas. Genetic syndromes such as neurofibromatosis and Li-Fraumeni cancer syndrome are associated with an increased risk of sarcoma.[10]
Malignant peripheral nerve sheath tumors are almost always seen in patients with neurofibromatosis type 1, an autosomal dominant syndrome that is related to an abnormality of a pericentromeric gene on chromosome 17. The product of this gene, neurofibromin, is a ubiquitous protein with tumor-suppressor activity.[11]
Li-Fraumeni syndrome is caused by a germline deletion affecting the tumor-suppressor gene p53 and may lead to a higher risk of rhabdomyosarcoma, early onset of breast cancer, and other tumors.[12] Some have suggested chronic lymphedema, physical agents such as ionizing radiation, and certain chemical agents, including phenoxyherbicides and chemotherapeutic agents, may be associated with an increased risk, but the evidence linking these to sarcoma is inconclusive and often contradictory.[13]
The nomenclature of soft tissue tumors is often confusing because these tumors represent a histologically heterogeneous group. Broadly, the most important distinction is between benign lesions, which are those that do not metastasize, although their local effects may cause significant morbidity or even death, and malignant lesions, which generally have the potential to metastasize, although a well- differentiated liposarcoma is an important exception.[14]
Well-differentiated liposarcomas very rarely metastasize; however, local recurrence can be a difficult problem if they are inadequately resected. Pathologic examination of a malignant tumor will determine its grade, which is an attempt to predict the probability of metastasis. Low-grade lesions have a less than 15% chance of metastasis and high-grade lesions have a greater than 15% chance of metastasis.[15]
Most soft tissue sarcomas are derived from a mesodermal cell origin, with the exception of the malignant peripheral nerve sheath tumors, which are neuroectodermal in origin. Soft tissue sarcomas are grouped according to their presumed cell of origin (e.g., liposarcoma from lipocyte cell line, rhabdomyosarcoma from primitive skeletal muscle cell line).
However, some soft tissue sarcomas, such as malignant fibrous histiocytoma, have no known cell of origin. Nevertheless, the biological activity of soft tissue sarcomas is similar for almost all cell types, and thus for purposes of diagnosis and management this group of tumors has historically been discussed as a whole,[10] although finer-grained approaches are becoming more feasible with better pathologic techniques.
Clinical assessment
All soft tissue masses should be approached with the awareness that they may represent a sarcoma. While there are no absolute clinical indicators that will distinguish benign from malignant soft tissue masses, several clinical features exist that will suggest the possibility of a malignancy to an alert clinician.
Failure to recognize these features at initial assessment can lead to misdiagnosis and interventions that may compromise survival or limb salvage opportunities once the correct diagnosis is made. For example, in one case a 68-year-old male being treated with anbicoagulants for abrial fibrillation presented with a soft tissue mass.
The mass was managed as a spontaneous hematoma with percutaneous drainage for several months. After the lesion became fungating (Figure 1), MR images were obtained. Subsequent biopsy confirmed malignant fibrous histiocytoma and the patient was referred to BCCA. A partial hemipelvectomy was performed (Figure 2) and systemic staging investigations identified metastatic lung lesions.
{kind=link}
{kind=link}
Most patients with a soft tissue sarcoma present with a painless mass, and few have systemic symptoms such as fever, weight loss, or malaise.[13] Though not common, pain or tenderness can occur; when it does, the possibility of malignancy is greater.
Rapidly increasing size, particularly in a lesion that was previously stable, suggests malignancy, as does the sudden appearance of a new large mass. Family history is usually negative, although patients should be asked about conditions such as neurofibromatosis. Exposure to industrial chemicals has not been conclusively proven to be associated with sarcoma.
Physical examination is the key to distinguishing benign from malignant soft tissue masses in the office (Figure 3). Lesions that are larger than 5 cm in any dimension, deep to the deep fascia, or that are growing rapidly should be assumed to represent soft tissue sarcomas until proven otherwise.
{kind=link}
In addition, superficial lesions that are smaller than 5 cm in any dimension but are adherent to the deep fascia or surrounding structures should be treated with suspicion. The consistency of a lesion can be misleading, as sarcomas can be quite soft and some benign lesions such as fibromatosis can be quite hard and irregular.
The relationship of a lesion to the deep fascia is often obvious on examination, but can be subtle. Lesions that feel superficial can be further evaluated by first attempting to mobilize the lesion with the patient relaxed, and then again with the underlying musculature tensed. If the lesion remains mobile, it is most likely not adherent or deep.
If the mass does not demonstrate any of these concerning features, serial examination every 3 to 6 months is sufficient management. Patients should also be instructed to return immediately if the mass becomes painful, tender, or starts to rapidly increase in size.
Systemic examination should not be neglected. While most sarcomas primarily metastasize to the lungs, some sarcomas, such as synovial sarcomas and epithelioid sarcomas, have a predilection for lymphatic metastasis, and the regional lymph nodes should be examined.
Lymphomas can present with soft tissue masses resembling sarcomas, and widespread lymphadenopathy may give a clue to this diagnosis. Evidence of other soft tissue masses should be sought, along with stigmata of genetic conditions such as neurofibromatosis (café-au-lait spots, axillary and groin freckling).
Chest and abdominal examination should be performed. Some sarcomas, such as high-grade myxoid liposarcomas and round cell liposarcomas, may metastasize to the abdomen, while clear cell sarcomas of soft parts may metastasize to the brain.
Investigations
A lesion that has not been rapidly increasing in size, is smaller than 5 cm, and is demonstrably superficial and does not adhere to the deep fascia has a low probability of representing a sarcoma. Further imaging for these lesions is of unclear benefit. An ultrasound examination can confirm the size of the lesion and its relationship to the fascia and is more readily available than MRI.
However, ultrasound images will not be able to give any reliable information on the underlying pathology and clincians should not be falsely reassured by ultrasound findings.[16,17] An ultrasound investigation is not a substitute for careful clinical examination and is not utilized for investigation of suspicious masses at the BC Cancer Agency.
If a lesion has features suggestive of a sarcoma, then more complete imaging is warranted. It is important to complete all imaging studies prior to any biopsy for two reasons.
First, it is impossible to accurately place a biopsy site so that it is consistent with future limb salvage procedures without detailed information about the lesion and its relationship to the normal anatomy that is now distorted by the mass. This may lead to an amputation being required in a limb that would otherwise have been salvageable.
Second, the biopsy itself causes edema and other changes that can interfere with interpretation of any subsequent imaging.
MRI is recognized as the standard imaging modality for large or deep soft tissue lesions. Gadolinium enhancement can improve the diagnostic accuracy.[8] As well as providing some information about the diagnosis, MR images give excellent anatomical detail to allow planning of the biopsy and surgery.
However, most lesions will still require some sort of tissue diagnosis regardless of what the MR images reveal. Although there can be a significant waiting period for MRI services in the community, the procedure can be obtained within 7 days by a subspecialist sarcoma surgeon in BC, and a referral for a suspicious mass should not be delayed while waiting for imaging services.
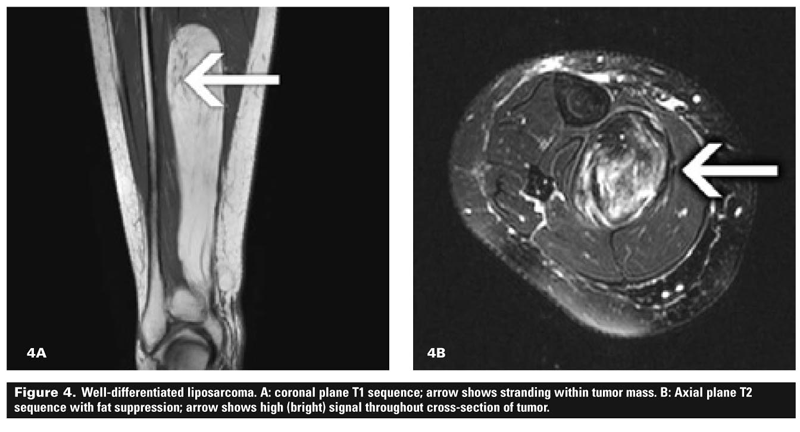
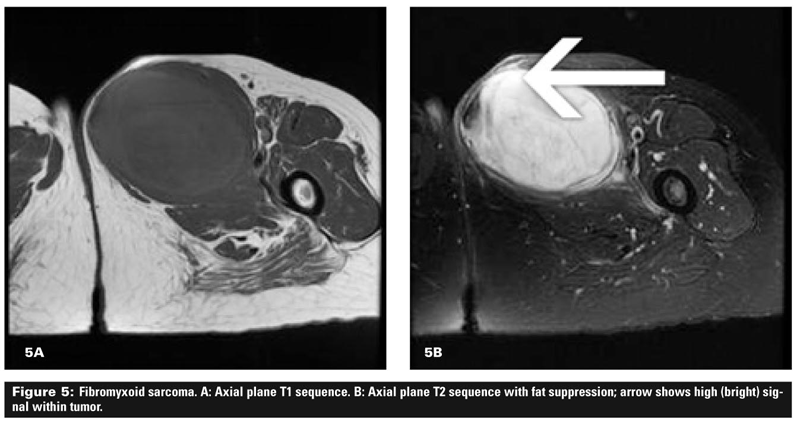
Figure 4 shows a well-differentiated liposarcoma that largely follows the signal of fat. The sarcoma is deep and large with some stranding (A) and areas of high signal (B), meaning that it is unlikely to be a lipoma. Figure 5 shows a fibromyxoid sarcoma, a high-grade lesion that is seen to be deep and large in the axial plane view (A) and also shows high signal (B) on the T2 sequences.
{kind=link}
{kind=link}
Plain radiographs are rarely helpful when investigating soft tissue masses, although lesions such as hemangiomas, synovial sarcomas, and lipomas can show intralesional calcification. Computed tomography is inferior to MRI but is sometimes used when the patient has a pacemaker or other condition precluding MRI, or where the lesion is near a large metallic implant that would generate a lot of magnetic interference.
PET scanning remains experimental for investigating soft tissue masses, but will most likely develop an important role in the management of soft tissue sarcoma because of its ability to provide information on the biological activity of the tissue. Bone scans have no role in the preoperative investigation of a soft tissue sarcoma.
If regional lymphadenopathy is detected clinically, it can be further evaluated with MRI. If the inguinal and pelvic nodes are involved, then a CT scan of the abdomen can reveal para-aortic lymphadenopathy. PET-CT scanning may be useful in distinguishing reactive from malignant lymphadenopathy, but requires great clinical experience and cannot be recommended for routine use.
Staging
Once the diagnosis of a soft tissue sarcoma has been made it is important to complete the staging process. In practice it is useful to think of staging as being local and systemic.
Local staging
Local staging assesses the extent of the primary tumor mass and gives information on its resectability as well as prognosis. The most useful imaging modality for local staging is MRI.
Optimally, this will be used prior to any biopsy, since local trauma from a biopsy can make interpretation of the MR images problematic. In selected cases of vascular compromise secondary to the sarcoma, local radiographs and vascular studies can also make up part of the local staging process.
Systemic staging
Systemic staging assesses metastatic disease, and can be delayed until after a biopsy has confirmed the diagnosis. Although soft tissue sarcomas predominantly metastasize to the lung, some, such as synovial sarcoma, have a predilection for lymphatic spread.
Imaging of the lungs with a chest X-ray or a CT scan is mandatory, and we recommend obtaining a baseline CT scan once the diagnosis of soft tissue sarcoma has been made. Lymphatic spread can sometimes be assessed clinically.
For this, added MRI or CT scanning can be helpful, combined with biopsy of nodes felt to possibly contain disease. Distinguishing malignant chest lesions from reactive changes may require serial imaging.
Several staging systems are in common use. The American Joint Committee on Cancer/International Union Against Cancer (AJCC/UICC) system is useful for prognosis, but difficult to administer. The Memorial Sloan Kettering (MSK) system is easier to administer and gives good prognostic information.
It relies on the accumulated number of adverse prognosticators: large tumor size (maximum diameter greater than 5 cm), deep location within the extremity, and high histologic grade. As the number of adverse factors increase, the probability of systemic metastasis occurring within 5 years also increases. If all three poor prognosticators are present, 5-year metastasis-free survival drops to approximately 48%.[18]
The surgical staging system reported by Enneking[15] is widely used by surgeons. It combines histological grade and compartmental status to predict outcome.
The concepts of compartmental status, tissues as barriers to tumor spread, and margin assessment described by Enneking are integral to the planning of tumor resection and assessment of limb salvage. Its usefulness for prognostication is more limited than the AJCC/ UICC or MSK systems, and most centres use a combination of these staging systems.
Biopsy
At BCCA we prefer all of our patients to be referred prior to having a biopsy performed. Biopsy should be considered following careful clinical assessment and review of the appropriate imaging studies.
If there is any doubt about whether or how a biopsy should be performed, the assistance of a surgeon with experience in the management of soft tissue sarcoma should be sought prior to undertaking any intervention on the lesion.[19,20]
Inappropriately planned or executed biopsy can result in misdiagnosis, increased mortality, and lost opportunities for limb salvage surgery.[1-4] Unfortunately, the incidence of inappropriate biopsy remains high even in Canada’s highly sophisticated health care system.
Core needle biopsy, usually with the assistance of image guidance, is our predominant practice. This provides information on the cells and the tissue architecture. The use of image guidance allows accurate needle placement and confirmation that lesional tissue has been obtained without contamination of the surrounding or deeper tissue.
Sufficient material can be obtained for a full pathologic assessment, which may involve an array of immunohistochemical stains.[20] The small scar generated also facilitates resection of the needle track en bloc with the sarcoma at the time of definitive surgery. However, some authors recommend a formal incisional biopsy as the best option in all cases.[19]
Fine needle aspiration is not useful in diagnosing sarcoma and we never recommend it. It reveals no infomation about the architecture of the tissue and provides very small amounts of material, making pathologic assessment difficult.
In addition, the needle track and extent of contamination is almost always untraceable, making its resection difficult at the time of definitive surgery.
Sarcomas are very transplantable lesions and all biopsy tracks must be resected at the time of definitive surgery, regardless of the biopsy technique used.
This means that the positioning of the biopsy should be in line with future limb salvage incisions (which are often not along standard surgical exposure lines); all biopsy incisions should be made in line with the longitudinal axis of the body part.
All biopsy tracks should be marked so they can be seen clearly. The track should not be placed close to major neurovascular structures because of the risk of contamination, but going through the centre of a muscular compartment can also cause considerable morbidity. Strict hemostasis must be obtained as a large hematoma may carry contaminated material some distance from the original lesion.
To reiterate, if sarcoma is in the differential diagnosis, consultation with or referral to the surgeon who will perform the definitive resection should be obtained prior to biopsy. Damron and colleagues suggest that a successful biopsy also requires the presence of a pathologist experienced in musculoskeletal tumors and a clinician able to interpret and manage the results of that biopsy appropriately.[21]
Management
Management of soft tissue sarcomas can be considered in terms of local and systemic control.
Local control
Local control is usually achieved with surgical resection, often combined with radiation therapy. Regardless of whether metastatic disease is present at the time of diagnosis, most patients with soft tissue sarcoma will still require local treatment in order to achieve symptomatic relief.
If left untreated, the primary tumor can grow extremely large, compromising neurovascular structures, and fungating through the skin.
Planning local control requires multidisciplinary consultation and a careful consideration of the anticipated margins and the implication of these for tumor recurrence and functional outcome. Enneking’s classification of margins as outlined in Table 1 should be considered.
{kind=link}
Note that an amputation does not necessarily represent a radical margin, as the line of resection may still have passed through the tumor. Most centres no longer aim for radical margins since radiation therapy combined with a wide margin resection can achieve a similar rate of local recurrence.[22]
In our practice at BCCA, if a very wide margin can be achieved with minimal morbidity for a small lesion, adjuvant radiotherapy may not be necessary. Some centres in Scandinavia suggest margins as small as 3 cm can be managed without radiotherapy with no increase in the local recurrence rates.[23]
These are typically small lesions located within large muscle groups or in the superficial tissues. Most lesions, however, will be resected with margins of less than 3 cm, and so will require adjuvant radiotherapy.
Some tissues, such as fascia, are resistant to tumor spread, and so the assessment of how much normal tissue represents a satisfactory margin in any individual case is a highly specialized decision, and one still under considerable debate.[20]
A wide margin is not always surgically achievable because of anatomical considerations. In areas such as the face or neck, radiation is relied upon to address the microscopically positive margin. In the extremities, a decision may have to be made between amputation and limb salvage based on the proximity or encasement of neurovascular structures.
Several papers support the combination of radiotherapy and careful removal of the tumor from nearby neurovascular structures, such as the sciatic nerve, using techniques such as epineural dissection.[24]
Although this technique may result in a microscopically positive margin, low recurrence rates have been observed with adjuvant or neoadjuvant radiotherapy.[25]
An intralesional or an unplanned microscopically positive margin is associated with high recurrence rates, despite adjuvant radiation therapy. For this reason, patients referred to us who have undergone previous surgery with such margins will receive preoperative radiation and a complete resection of the previous surgical bed if this is feasible.
Even with this aggressive treatment, these patients still have inferior outcomes, underscoring the importance of appropriate diagnosis and management by the initial treating physician.
Radiation therapy is considered for all patients with soft tissue sarcoma. Although radiation therapy can be given preoperatively or postoperatively, a Canadian prospective randomized trial found preoperative radiotherapy to be associated with better long-term functional outcomes.[26]
In this study the preoperative group had similar recurrence rates to the postoperative group, but the preoperative group had fewer long-term radiation effects, particularly when radiation was used in the lower limbs. However, the preoperative radiotherapy group had a much higher wound complication rate.
Our centre favors preoperative radiotherapy based on the fact that short-term wound problems are surmountable (all patients in the trial eventually had healed wounds) and preferable to the long-term irreversible functional problems associated with the larger radiation fields required for postoperative radiation therapy.
However, this remains controversial in some centres, and this trial has been criticized because its primary outcome was local recurrence, not functional outcome.
Systemic control
Systemic control of soft tissue sarcoma can involve surgery, radiotherapy, chemotherapy, or a combination of all three. Lymphatic metastases can be cleared with surgical resection combined with radiation therapy if the involvement is not too extensive.
Resection of lung metastases that are few in number, small, and peripherally placed has been associated with improved prognosis and even cure.
The use of chemotherapy remains controversial. Patients who have large, high-grade, deep lesions based in the extremity and no evidence of systemic disease at presentation may benefit from chemotherapy, both in terms of survival and time to recurrence.[27]
This benefit, however, is small. In our institution each patient is assessed on an individual basis, taking into account their ability to tolerate the toxicity of chemotherapy.
Conclusions
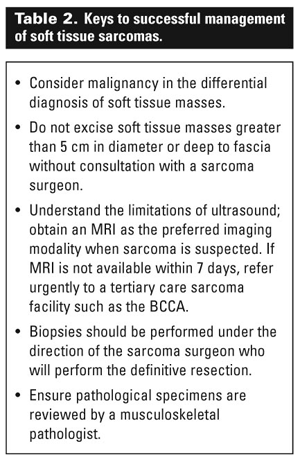
Distinguishing a soft tissue sarcoma from the many benign soft tissue masses that present remains a challenge for the primary care physician. Yet careful management of soft tissue masses is imperative to optimize the outcome of the relatively few sarcomas present (Table 2).
{kind=link}
Errors in the early diagnosis and management of these lesions are associated with poorer outcomes for these patients. Unfortunately, these errors still occur, even in modern health care systems, as demonstrated by Paszat and colleagues in Ontario.[28]
Masses that should raise suspicion are those with the following characteristics:
• Larger than 5 cm.
• Located deep to the deep fascia.
• Rapidly growing.
• Painful or tender.
Advice from, or referral to, a centre with expertise in the multidisciplinary management of soft tissue sarcoma should be considered prior to any intervention on these lesions, even biopsy.
Interventions on small superficial lesions should be made through longitudinal incisions and the deep fascia should be left intact to facilitate future resection of the surgical bed in case a sarcoma is diagnosed. All resected specimens should be examined by a musculoskeletal pathologist.
The current practice of the BCCA soft tissue sarcoma group is to refer patients with benign masses back to the referring surgeon for further management.
Most errors in the early management of soft tissue sarcoma arise from failure to consider sarcoma in the differential diagnosis rather than from lack of knowledge or skill. Obtaining inadequate specimens can lead to falsely reassuring pathology reports if the clinician is not vigilant.
Pre- referral intervention on large soft tissue masses without adequate workup does occur in BC, with sometimes disastrous results for the patient, but the majority of soft tissue sarcomas in our practice are appropriately referred.
The BC Cancer Agency soft tissue sarcoma group is willing to review any case where soft tissue sarcoma is in the differential diagnosis.
Competing interests
None declared.
References
1. O’Sullivan B, Pisters PW. Staging and prognostic factor evaluation in soft tissue sarcoma. Surg Oncol Clin N Am 2003; 12:333-353.
2. Mankin HJ, Mankin CJ, Simon MA. The hazards of biopsy revisited. Members of the Musculoskeletal Tumor Society. J Bone Joint Surg Am 1996;78:656-663.
3. Simon MA, Blermann JS. Biopsy of bone and soft-tissue lesions. J Bone Joint Surg Am 1993;75:616-621.
4. Noria S, Davis A, Kandel R, et al. Residual disease following unplanned excision of soft-tissue sarcoma of an extremity. J Bone Joint Surg Am 1996;78:650-655.
5. Canadian Institute for Health Information. Supply, Distribution and Migration of Canadian Physicians. Ottawa: CHI; 2005:60.
6. National Cancer Institute. Cancer Facts. NCI; Bethesda, MA: 2000.
7. Zahm SH, Fraumeni JF. The epidemiology of soft tissue sarcoma. Semin Oncol 1997;24:504-514.
8. Singer S, Eberlein TJ. Surgical management of soft tissue sarcoma. In: Cameron JL, Balch CM, Langer B, et al. (eds). Advances in Surgery. Vol. 31 St. Louis: Mosby-Year Book, Inc.; 1997:395-420.
9. Rydholm A, Berg NO, Gullberg B, et al. Epidemiology of soft-tissue sarcoma of the locomotor system. A retrospective population-based study of the inter- relationships between clinical and morphologic variables. Acta Pathol Microbiol Immunol Scand [A] 1984;92:363-374.
10. Lewis JJ, Brennan MF. Soft tissue sarcomas. Curr Probl Surg 1996;33:817-872.
11. DeClue JE, Cohen BD, Lowy DR. Identification and characterization of the neurofibromatosis type 1 protein product. Proc Natl Acad Sci USA 1991;88:9914-9918.
12. Olivier M, Goldgar DE, Sodha N, et al. Li-Fraumeni and related syndromes: Correlation between tumour type, family structure, and TP53 genotype. Cancer Res 2003;63:6643-6650.
13. Posner MC, Brennen MF. Soft tissue sarcomas. In: Holleb AI, Fink DJ, Murphy GP (eds). Textbook of Clinical Oncology. Atlanta, GA: American Cancer Society; 1991: 359-376.
14. Deyrup AT, Weiss SW. Grading of soft tissue sarcomas: The challenge of providing precise information in an imprecise world. Histopathology 2006;48:42-50.
15. Enneking WF, Spanier SS, Goodman MA. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop Relat Res 1980;153:106-120.
16. Doyle AJ, Miller MV, French JG. Ultrasound of soft-tissue masses: Pitfalls in interpretation. Australas Radiol 2000; 44:275-280.
17. Brouns F, Stas M, De Wever I. Delay in diagnosis of soft tissue sarcomas. Eur J Surg Onco 2003;29:440-445.
18. Wunder JS, Healey JH, Davis AM, et al. A comparison of staging systems for localized extremity soft tissue sarcoma. Cancer 2000;88:2721-2730.
19. Ham SJ, Van der Graf WTA, Pras E, et al. Soft tissue sarcoma of the extremities. A multimodality diagnostic and therapeutic approach. Cancer Treat Rev 1998;24: 373-391.
20. Aboulafia A. Biopsy. Instr Course Lect 1999;48:587-590.
21. Damron T, Beauchamp C, Rougraff B, et al. Soft-tissue lumps and bumps. J Bone Joint Surg Am 2003;85A:1142-1155.
22. Rosenberg SA, Tepper J, Glatstein E, et al. The treatment of soft-tissue sarcomas of the extremities: Prospective randomized evaluations of (1) limb-sparing surgery plus radiation therapy compared with amputation and (2) the role of adjuvant chemotherapy. Ann Surg 1982;196: 305-315.
23. Rydholm A. Surgery without radiotherapy in soft tissue sarcoma. Acta Orthop Scand Suppl 1997;273:117-119.
24. Clarkson PW, Griffin AM, Catton CN, et al. Epineural dissection is a safe technique that facilitates limb salvage surgery. Clin Orthop Relat Res 2005; 438:92-96.
25. Gerrand CH, Wunder JS, Kandel RA, et al. Classification of positive margins after resection of soft-tissue sarcoma of the limb predicts the risk of local recurrence. J Bone Joint Surg 2003;83B:1149-1155.
26. O’Sullivan B, Davis AM, Turcotte R, et al. Preoperative versus postoperative radiotherapy in soft-tissue sarcoma of the limbs: A randomised trial. Lancet 2002; 359:2235-2241.
27. O’Byrne K, Steward WP. The role of chemotherapy in the treatment of adult soft tissue sarcomas. Oncology 1999; 56:13-23.
28. Paszat L, O’Sullivan B, Bell R, et al. Processes and outcomes of care for soft tissue sarcoma of the extremities. Sarcoma 2002;6:19-26.
Dr Pike is a resident in the Department of Orthopaedics at the University of British Columbia. Dr Clarkson is a clinical instructor in the Department of Orthopaedics at UBC, and an orthopaedic oncologist at the BC Cancer Agency. Dr Masri is a professor in and head of the Department of Orthopaedics at UBC, and an orthopaedic oncologist at the BC Cancer Agency.