The long QT syndrome: Half a century of electrophysiology
Issue: BCMJ,
vol. 44, No. 2, March 2002,
Pages 86-92 Clinical Articles
Recognized more than 50 years ago, the long QT syndrome is a group of cardiac ion channel abnormalities caused by mutations in the genes encoding them. The clinical manifestations include syncope and sudden death in young individuals due to ventricular arrhythmias. This article reviews some of the developments in our understanding of this primary electrical disease, which remains an important treatable cause of sudden death and syncope in children and young adults.
A review of the salient features and recent developments regarding the long QT syndrome, a group of cardiac ion channel abnormalities caused by mutations in the genes encoding them.
Nearly 45 years have passed since Jervell and Lange-Nielsen described four of six siblings, three of whom died, with congenital deafness, fainting spells, and prolongation of the QT interval on electrocardiogram (ECG).[1] A few years later, Romano[2] and Ward[3] reported similar patients but with normal hearing. In 1950, an eminent cardiologist wrote of the electrocardiograms demonstrating long QT syndrome: “I am afraid you will have to place [the electrocardiograms] in a group called ‘screwballs.’”[4] Since these important descriptions, our knowledge about the long QT syndrome has grown exponentially. Long QT syndrome is now known as a familial cardiovascular disorder characterized by abnormal cardiac repolarization, seen as a prolonged QT interval on the surface ECG, caused by mutations in the genes that encode for ion channels.[5-8] Syncope and sudden death occur in young individuals due to ventricular arrhythmias, primarily torsade de pointes and ventricular fibrillation.
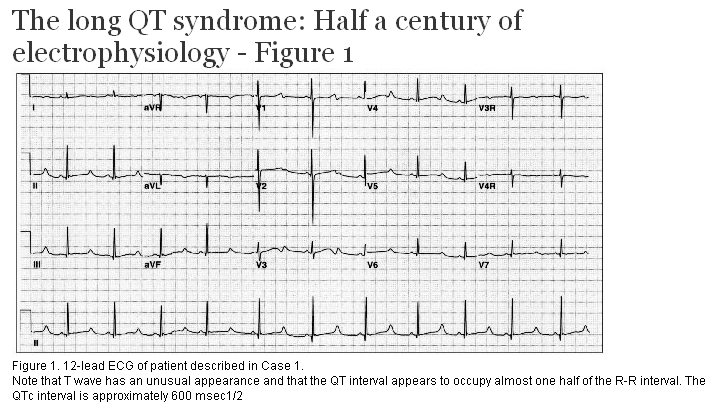
A previously well 8-year-old girl was swimming with several friends in a neighbor’s pool. Suddenly, she sank to the bottom of the pool and remained motionless. Initially thought to be at play, she was soon rescued from the pool and found to be unresponsive. She received bystander CPR and regained consciousness rapidly. She was transferred to our institution for evaluation. The family history revealed that her mother had been diagnosed with a seizure disorder in her late teens. The patient’s ECG is shown in Figure 1. A diagnosis of long QT syndrome (LQTS) was made in the patient, her mother, and her brother. She remains asymptomatic on beta-blockers.
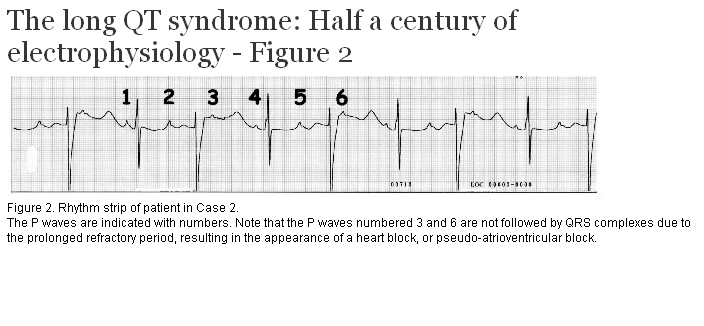
A 30-year-old primigravida was in for a routine antenatal visit at 30 weeks gestation. The fetal heart rate was noted to be slow, so a fetal echocardiogram was arranged. This confirmed fetal bradycardia but did not demonstrate any structural abnormality. The presumptive diagnosis was congenital heart block and the fetus was monitored closely. The fetus continued to grow normally, with no evidence of hydrops. The fetal heart rate was approximately 100 beats per minute, but demonstrated some variability. The pregnancy was carried to term and a baby girl was delivered. The baby was noted to be persistently bradycardic. Her rhythm strip is shown in Figure 2. A diagnosis of long QT syndrome with pseudo-atrioventricular block was made. In the first few hours of life, she developed non-sustained ventricular tachycardia. She received a pacemaker and was started on beta-blockers. She has been growing and developing normally since that time. She has not had further arrhythmias.
Genetics and molecular biology
In the current era, no discussion of long QT syndrome is meaningful without familiarity with the molecular biology of the condition. Traditionally, the modes of inheritance were considered as autosomal dominant, as described by Romano[2] and Ward,[3] in which there is not usually any other phenotypic abnormality; and autosomal recessive, as in the Jervell and Lange-Nielsen[1] description. The latter is characterized by the presence of sensorineural congenital deafness, due to the same ion channel abnormality.[9] There are also sporadic cases and acquired cases. With advances in molecular biology, LQTS is no longer thought of as simply the Jervell and Lange-Nielsen and Romano-Ward syndromes. Rather, LQTS is a so-called channelopathy, a disease due to abnormalities in the ion channels controlling the action potential in electrically active cells—in this case, the myocardial cells. Ion channels are large glycoprotein molecules spanning cell membrane and forming pores. These channels generally consist of several transmembrane regions and multiple subunits, which coassemble to give rise to the functional channel. Pores have permeability and selectivity, the control of which is maintained in highly preserved sequences of amino acids.[6]
A simplified action potential is illustrated in Figure 3. The first phase (phase 0) of the action potential is mediated by a rapid influx of sodium through sodium channels into the cell. This is seen on the surface ECG as the QRS complex. This is followed by activation of a number of potassium currents, the function of which is to return the cell to its resting potential. This is countered by the inflow of calcium ions resulting in the plateau phase of the action potential (phase 2). Subsequently, as calcium currents subside, the cell is repolarized by the potassium currents (phase 3), giving rise to the T wave on ECG. Several excellent descriptions of the cardiac action potential exist in the literature.[8,10,11] Note that very small alterations in current flow can have dramatic electrophysiologic consequences.
In 1990, a DNA marker was linked to the occurrence of long QT syndrome in a large family with several members affected by the disease.[12] During the next several years, the search for the gene(s) responsible for LQTS took place, with identification of three genes.[13-16] Since then, over 200 mutations in six genes have been identified. LQTS is considered in subgroups, according to the molecular biology of the ion channels (Table 1). The potassium channel defects result in a decrease in function, resulting in prolongation of the repolarization phase. In LQT3, the SCN5A mutation does not allow complete inactivation of sodium inflow, allowing continued entry into the cell, prolonging repolarization. At present, this is the only defect resulting in a gain of function in the LQTS, having implications for treatment options (see below).
Another advance came when a patient with the Jervell and Lange-Nielsen syndrome, an autosomal recessive form of long QT syndrome, was found be homozygous for mutations ascribed to the autosomal dominant form of the disease.[9] The hearing loss was found to be due to complete loss of the IKs potassium current and is a recessive trait.[9] While the parents of offspring with Jervell and Lange-Nielsen should have Romano-Ward syndrome, this has recently been questioned.[17]
From genetic defect to clinical manifestations
The role of the autonomic nervous system in clinical events has long been recognized.[7,18,19] This is apparent in patients who experience events during physical activity or during intense emotions. Now we understand that the role of the autonomic nervous system in the development of clinical events is related to the effects it has on ion channels and currents. Simplistically, the effect of the mutations identified thus far is to introduce an excess of positive charges into the cell.[10] This can result in abnormal secondary depolarizations known as early afterdepolarizations. A rapid rate followed by a pause, such as is seen with a premature ventricular complex, favors the formation of early afterdepolarizations.[10] Since ion currents are distributed inhomogenously in the myocardium, regional differences in action potential behavior can set up re-entrant arrhythmias. This is believed to be the case with torsade de pointes in LQTS.
Long QT syndrome is estimated to occur in 1 in 7000 to 10,000 individuals.[6,7] In order to delineate the spectrum of LQTS further, a prospective international registry was initiated in 1979.[20] This registry has provided a wealth of clinical information and has fed the extensive molecular biology in the field. Now, with a large number of patients genotyped, specific patterns are being related to the underlying mutations.[7,21]
The clinical manifestations are broad and range from the asymptomatic carrier to sudden death. Typical symptoms include palpitations, syncope, and seizures, although cardiac arrest may be the initial presentation. The occurrence of syncope during times of emotional excitement, physical exertion, or with auditory stimuli is very suggestive of long QT syndrome. Females have longer QT intervals and are more prone to torsade after puberty.[7] There are patients who are diagnosed with vasovagal episodes or seizure disorders before the repolarization abnormality is recognized, such as the mother in Case 1. Naturally, a family history of sudden death should be sought.
The most important finding is that of a prolonged QT interval on the resting ECG. Since prolongation of the QT interval can be observed in many conditions, including electrolyte disturbances—most notably hypocalcemia and hypomagnesemia, pharmacologic agents, central nervous system disorders, and heart disease (Table 2)—a careful workup must exclude these causes. In long QT syndrome, the T wave is more likely to be biphasic or notched, and certain ECG patterns have been associated with certain genotypes. ECGs should be obtained from the immediate family, although approximately 40% of new cases are sporadic. However, 5% to 10% of both genotyped and symptomatic family members have a normal QT interval.[7,22]
Due to the difficulty in making a diagnosis in some patients, Schwartz proposed a scoring system combining ECG and clinical data (Table 3).[23] While this is helpful in some cases, it does little to sort out the patients with a borderline prolongation of the QTc. In most patients a 12-lead ECG suffices; however, in some patients additional testing will increase the likelihood of making the correct diagnosis. Sometimes, several ECGs are required before recording the prolonged QT interval. The QTc as derived by Bazett’s formula is generally used, as determined by dividing the measured QT by the square root of the R-R’ interval (QTc=QT/√R-R’). Normal values should be less than 0.46 msec1/2 beyond the first week of life.
Holter monitoring is sometimes useful in detecting prolongation of the QT interval and T wave changes. The T wave may change in amplitude and in direction from beat to beat, a phenomenon known as T wave alternans. The Holter monitor is also an important test for detecting occult arrhythmias. Sinus bradycardia is common, even without beta-blockade, and has been recognized in the fetus. The presentation may be that of profound bradycardia due to extreme prolongation of the QTc leading to pseudo heart block, such as that described in Case 2.
Exercise testing is often used to document peak heart rate, inducibility of arrhythmias, and to determine the QT interval in the recovery phase of the test, where QT prolongation may be most marked. Pharmacologic and electrolyte challenges are also done in some cases in the electrophysiology lab.
At this point, genetic testing has not reached a clinical phase. Arrangements can be made with some of the large research labs, particularly in the United States, but this testing has not evolved to a diagnostic service.
The newborn found to have prolongation of the QTc interval on ECG presents a particular challenge. Villain reported the outcomes of 15 selected newborns with QTc prolongation and found that in one-third the QTc normalized. Those with a QTc <500 msec1/2 with no ventricular arrhythmias or heart block on day 4 had a benign course.[24] In 1998, Schwartz et al. published the results of a formidable study that lasted almost 20 years and involved the follow-up of 33,034 of 34,442 neonates who had ECGs on the third or fourth day of life.[25] Of the 24 infants who died of the sudden infant death syndrome (SIDS), 12 had QTc >440 msec1/2, and the suggestion was that a prolonged QTc on the third or fourth day of life carries with it a high risk of succumbing to SIDS. There was much discussion in response to this paper and the association of long QT syndrome and SIDS.[26-28] The paper did not draw attention to the low positive predictive value of QTc prolongation, nor were those at highest risk for SIDS included, namely premature and critically ill infants. The authors have since described a case in which the clinical and molecular diagnosis of LQTS was made in a patient surviving a near-miss episode that had the features of SIDS.[29] It is very likely that some deaths ascribed to SIDS are due to cardiac arrhythmias. Presently, though, what proportion and, more importantly, how to pre-empt them, remain unknown.
While many drugs may prolong the QT interval, the vast majority are not associated with lethal ventricular arrhythmias. An example of this is the gastric motility agent cisapride, now off the market due to reported cardiac deaths. Most arrhythmias appear to relate to drug levels, often in the setting of medication interactions.[30,31] However, it has been demonstrated that some patients with drug-induced torsade de pointes may be silent carriers of genetic defects affecting ion channel function. The addition of the drug unmasks the molecular defect and results in clinical disease. Many of the culprit drugs appear to affect the HERG IKr channel. It has been suggested that its relatively large pore size and structure allow the drug-channel interaction.[8] Cisapride blocks the IKr current.[32] The list of drugs to avoid in the congenital long QT syndrome is extensive, and can be found at various web sites (e.g., www.qtdrugs.org).
The untreated mortality of long QT syndrome has been estimated to be >50% over 10 years. With treatment this figure is reduced approximately tenfold.[7] While the frequency of events is higher in LQT1 and LQT2 and in patients with longer QTc, the mean QTc and the lethality of events are highest in LQT3.[21]
Beta-blockers form the mainstay of therapy in long QT syndrome. The mortality has been significantly reduced using this modality of therapy alone.[7,19,33] With the recognition of LQTS subtypes, we may see the emergence of mutation specific treatments.[21,34] For example, the treatment of patients with sodium channel defects (LQT3) with the sodium channel blocking agent flecainide has shown some promise in a small group of patients.[34]
In some patients, pacing may be beneficial. This should be viewed as an adjunctive therapy and pacing these patients may require complex algorithms to eliminate the post-extrasystolic pauses that may lead to torsade de pointes.[10,19] Withdrawal of beta-blockers when pacing was instituted resulted in a high incidence of sudden death in the registry.[7]
Prior to the discovery of the channel defects underlying long QT syndrome, a prominent hypothesis was that sympathetic imbalance was at least partly responsible for the disease.[18,35] This was supported by the fact that stimulation of the left stellate ganglion produced prolongation of the QT interval and that beta-blockers significantly reduced symptoms. In a small number of patients who remained symptomatic despite beta-blockade, left cardiac sympathetic denervation has been performed. This resulted in a reduction of symptoms in the vast majority of patients.[7,35]
In recent years the use of the implantable cardioverter defibrillator (ICD) has grown exponentially. ICDs have been used effectively in patients with long QT syndrome. The use of ICD therapy should be approached very cautiously in these patients, who may have self-limited runs of arrhythmias and may receive many shocks. These, in turn, may precipitate further catecholamine release and lead to further clinical deterioration. The numbers of LQTS patients who have received this therapeutic modality is not large and patients who are refractory to other therapies may benefit.[7]
As with many other fields of medicine in which the scientific advances raise more questions, genotyping in long QT syndrome is teaching us some difficult lessons. Patients, including symptomatic patients, may have the LQTS gene but manifest a normal QTc on ECG. The distribution for carriers and noncarriers overlap significantly, particularly in the range of 0.44 and 0.47 sec. There is tremendous genetic heterogeneity in the syndrome and many patients with LQTS do not have mutations of the hitherto described genes. The penetrance (the manifestation of the disease in the presence of the gene) may not be as high as originally proposed. Recently, a family was described in whom the pattern of the Romano-Ward syndrome was inherited in an autosomal recessive fashion.[36] The different mutations may respond differently to different therapies, but most patients are not genotyped.
Long QT syndrome represents an area where the gap between basic science and clinical practice is narrowing. This will undoubtedly help us to understand the disease better and therefore treat our patients more effectively. However, despite our understanding of this primary electrical cardiac disease, it remains an important treatable cause of sudden death and syncope in children and young adults.
Table 1. Overview of long QT syndrome subtypes.
|
Chromosome |
Gene |
Ion current affected |
Relative frequency in genotyped |
Phenotype triggers |
Phenotype ECG |
|
|
LQTS 1 |
11p15.5 |
KCNQ1 (KVLQT1) |
Potassium |
~45% |
Activity, emotions |
Smooth, broad-based T waves |
|
LQTS 2 |
7q35-36 |
HERG |
Potassium |
~45% |
Emotions, auditory, activity |
Notched low-amplitude T waves |
|
LQTS 3 |
3p21-24 |
SCN5A |
Sodium |
~5% |
Sleep |
Late onset of T wave |
|
LQTS 4* |
4q25-27 |
Unknown |
Unknown |
Unknown |
Unknown |
Sinusoidal T wave |
|
LQTS 5 |
21q22 |
KCNE1 |
Potassium |
~2% |
Unknown |
Unknown |
|
LQTS 6 |
21q22 |
KCNE2 |
Potassium |
~2% |
Unknown |
Unknown |
* The gene product for LQTS 4 has not yet been identified.
Table 2. Acquired causes of QT prolongation.
|
Cardiovascular Pharmacologic Electrolyte Neurologic |
Table 3. Diagnostic criteria for long QT syndrome.
|
ECG findings* A. ≥480 msec1/2 Clinical history A. Syncope† Family history‡ |
Points 3
2
1 |
* In absence of medications or disorders known to affect these ECG findings
† Mutually exclusive
‡ Same family member cannot be counted in A and B
Scoring: ≤1: low probability; 2 – 3: intermediate; ≥4: high probability
The author would like to thank Drs Derek Human, John Yeung, and Jim Potts for their assistance with the preparation of this manuscript.
None declared.
Dr Sanatani will provide a more detailed reference list upon request; contact him at sanatani@interchange.ubc.ca.
References
1. Jervell A, Lange-Nielsen F. Congenital deaf-mutism, functional heart disease with prolongation of the Q-T interval, and sudden death. Am Heart J 1957;54:59-68.
2. Romano C, Gemme G, Pongiglione R, et al. Rare Dell’eta’ Pediatrica. Clinica Pediatrica 1963;45:656-683.
3. Ward OC. A new familial cardiac syndrome in children. J Irish Med Assoc 1964;54:103-106.
4. Levine SA, Woodworth CR. Congenital deaf-mutism, prolonged QT interval, syncopal attacks, and sudden death. N Engl J Med 1958;259:412-417.
5. Roden DM, Lazzara R, Rosen M, et al. Multiple mechanisms in the long-QT syndrome. Current knowledge, gaps, and future directions. The SADS Foundation Task Force on LQTS. Circulation 1996;94:1996-2012. PubMed Abstract Full Text
6. Ackerman MJ. The long QT syndrome: Ion channel diseases of the heart. Mayo Clin Proc 1998;73:250-269. PubMed Abstract
7. Schwartz PJ, Priori SG, Napolitano C. The long QT syndrome. In: Zipes DP, Jalife J (eds). Cardiac Electrophysiology: From Cell to Bedside. Philadelphia: W.B. Saunders, 2000:597-615.
8. Keating MT, Sanguinetti MC. Molecular and cellular mechanisms of cardiac arrhythmias. Cell 2001;104:569-580. PubMed Citation
9. Neyroud N, Tesson F, Denjoy I, et al. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat Genet 1997;15:186-189. PubMed Abstract
10. Viskin S. Long QT syndromes and torsade de pointes. Lancet 1999;354:1625-1633. PubMed Abstract
11. Rudy Y. Ionic mechanisms of cardiac electrical activity. In: Zipes DP, Jalife J (eds). Cardiac Electrophysiology: From Cell to Bedside. Philadelphia: W.B. Saunders, 2000:257-265.
12. Keating M, Atkinson D, Dunn C, et al. Linkage of a cardiac arrhythmia, the long QT syndrome, and the Harvey ras-1 gene. Science 1991;252:704-706. PubMed Abstract
13. Jiang C, Atkinson D, Towbin JA, et al. Two long QT syndrome loci map to chromosomes 3 and 7 with evidence for further heterogeneity. Nat Genet 1994;8:141-147. PubMed Abstract
14. Curran ME, Splawski I, Timothy KW, et al. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 1995;80:795-803. PubMed Abstract
15. Wang Q, Shen J, Splawski I, et al. SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 1995;80:805-811. PubMed Abstract
16. Wang Q, Curran ME, Splawski I, et al. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat Genet 1996;12:17-23. PubMed Abstract
17. Chen Q, Zhang D, Gingell RL, et al. Homozygous deletion in KVLQT1 associated with Jervell and Lange-Nielsen syndrome. Circulation 1999;99:1344-1347. PubMed Abstract Full Text
18. Schwartz PJ, Periti M, Malliani A. The long Q-T syndrome. Am Heart J 1975;89:378-390. PubMed Abstract
19. Eldar M, Griffin JC, Van Hare GF, et al. Combined use of beta-adrenergic blocking agents and long-term cardiac pacing for patients with the long QT syndrome. J Am Coll Cardiol 1992;20:830-837. PubMed Abstract
20. Moss AJ, Schwartz PJ, Crampton RS, et al. The long QT syndrome: A prospective international study. Circulation 1985;71:17-21. PubMed Abstract
21. Zareba W, Moss AJ, Schwartz PJ, et al. Influence of genotype on the clinical course of the long-QT syndrome. International Long-QT Syndrome Registry Research Group. N Engl J Med 1998;339:960-965. PubMed Abstract Full Text
22. Priori SG, Napolitano C, Schwartz PJ. Low penetrance in the long-QT syndrome: Clinical impact. Circulation 1999;99:529-533. PubMed Abstract Full Text
23. Schwartz PJ, Moss AJ, Vincent GM, et al. Diagnostic criteria for the long QT syndrome. An update. Circulation 1993;88:782-784. PubMed Citation
24. Villain E, Levy M, Kachaner J, et al. Prolonged QT interval in neonates: Benign, transient, or prolonged risk of sudden death. Am Heart J 1992;124:194-197. PubMed Abstract
25. Schwartz PJ, Stramba-Badiale M, Segantini A, et al. Prolongation of the QT interval and the sudden infant death syndrome. N Engl J Med 1998;338:1709-1714. PubMed Abstract Full Text
26. Hoffman JI, Lister G. The implications of a relationship between prolonged QT interval and the sudden infant death syndrome. Pediatrics 1999;103:815-817. PubMed Citation
27. Lucey JF. Comments on a sudden infant death article in another journal. Pediatrics 1999;103:812. PubMed Citation
28. Berul CI. Neonatal long QT syndrome and sudden cardiac death. Prog Pediatr Cardiol 2000;11:47-54. PubMed Citation
29. Schwartz PJ, Priori SG, Dumaine R, et al. A molecular link between the sudden infant death syndrome and the long-QT syndrome. N Engl J Med 2000;343:262-267. PubMed Citation Full Text
30. No authors listed. Lessons from cisapride. CMAJ 2001;164:1269,1271. PubMed Citation Full Text]
31. Sukkari SR, Sasich LD. Cisapride and patient information leaflets. CMAJ 2001;164:1276-1279. PubMed Citation Full Text
32. Walker BD, Singleton CB, Bursill JA, et al. Inhibition of the human ether-a-go-go-related gene (HERG) potassium channel by cisapride: Affinity for open and inactivated states. Br J Pharmacol 1999;128:444-450. PubMed Abstract
33. Viskin S. Cardiac pacing in the long QT syndrome: Review of available data and practical recommendations. J Cardiovasc Electrophysiol 2000;11:593-600. Pubmed Abstract
34. Schwartz PJ, Priori SG, Locati EH, et al. Long QT syndrome patients with mutations of the SCN5A and HERG genes have differential responses to Na+ channel blockade and to increases in heart rate. Implications for gene-specific therapy. Circulation 1995;92:3381-3386. PubMed Abstract Full Text
35. Schwartz PJ, Locati EH, Moss AJ, et al. Left cardiac sympathetic denervation in the therapy of congenital long QT syndrome. A worldwide report. Circulation 1991;84:503-511. PubMed Abstract
36. Priori SG, Schwartz PJ, Napolitano C, et al. A recessive variant of the Romano-Ward long-QT syndrome? Circulation 1998;97:2420-2425. PubMed Abstract Full Text
Shubhayan Sanatani, MD, FRCPC
Dr Sanatani is a pediatric cardiologist and electrophysiologist in the Division of Cardiology at British Columbia’s Children’s Hospital and an assistant professor in the Department of Pediatrics at the University of British Columbia.

{kind=link}
{kind=link}
{kind=link}